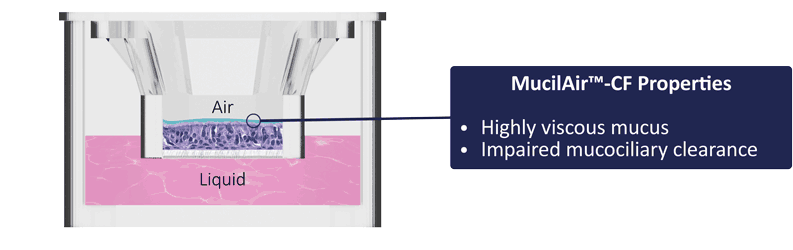
Epithelix has a large collection of CF cells with well characterized genotypes established by certified laboratories. MucilAir™-CF replicates the major phenotypes of CF diseases: absence of chloride current, impaired mucociliary clearance, etc. Epithelix possesses the most advanced equipments and expertise for studying the Cystic Fibrosis disease.
Epithelix provides the following services :
- Evaluation of Corrector, Activator or Potentiator of mutated CFTR on human airway epithelium from Cystic Fibrosis donors (MucilAir™-CF)
- Evaluation of the mucociliary clearance restoration on human airway epithelium from Cystic Fibrosis donors (MucilAir™-CF)
- In vitro inhibition of airway mucosa liquid absorption

 Mucociliary clearance on MucilAir™ Healthy
Mucociliary clearance on MucilAir™ Healthy